MEREDITH
A novel multiplatform data integration approach that identifies similarities among 4,434 patients taken from the Cancer Genome Atlas (TCGA) across 19 cancer types based on four genome-wide measurements platforms
Background
The use of genome-wide data in cancer research has become a standard approach to discover biomarkers for improved cancer subtyping. Applications in therapy response, prognosis prediction, and drug development are based on identifying groups of patients with similar molecular characteristics. To keep improving in these applications and thereby better characterize an individual patient’s cancer type, the trend is to move from single genome-wide measurements in a single cancer towards multiple same-sample genome-wide measurements across multiple cancers. Although current approaches shed light on molecular characteristics of various cancer types globally, the exact relationship between patients and the substructures that exists within cancer clusters is unclear. Results: We propose a novel genome-wide multiplatform integration approach (MEREDITH) that generates a two-dimensional landscape of all cancer samples emphasizing their local relationships. We integrated 4674 samples across 19 cancer types, derived from the cancer genome atlas (TCGA), containing gene expression (GE, n=18882), DNA-methylation (ME, n=11429), copy number variation (CN, n=23638) and microRNA expression (MIR, n=467) data. Cluster analysis revealed 18 clusters: eight clusters showed exclusive enrichment of one cancer type; five clusters were subtypes of one cancer type; and three clusters showed a complex collection of cancer types, i.e., the squamous cancers, colorectal cancers, and a novel grouping of kidney cancers. 1.37% of all cancer patients (n=64) are located outside their tissue-of-origin cluster from which specific subset of patients appeared to be associated with significantly worse overall survival. Systematic quantification of genome-wide platform contributions show that some substructures are driven by specific (combinations of) molecular features, whilst others are uniquely determined by the integration of all molecular feature types.
Conclusion
Taken together, MEREDITH integrates multiple genome-wide data exploiting the joint behavior of the different molecular features, produces sample distributions that better reflect the cancer type of origin, and supports visual exploration of the data and inspection of the contribution of the different genome-wide platforms. In short, a powerful tool to explore multiple platform pan-cancer data.
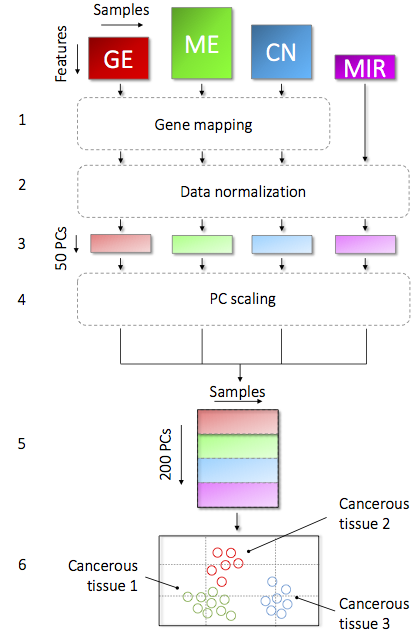
MEREDITH: a multiplatform data integration approach
We devised a novel multiplatform data integration approach (MEREDITH) to identify similarities among 4,672 patients taken from the Cancer Genome Atlas (TCGA7) across 19 cancer types based on four genome-wide measurements platforms: gene expression (GE, 18,882 features), DNA-methylation (ME, 11,429 features), copy number variation (CN, 23,638 features) and microRNA expression (MIR, 467 features) data. MEREDITH is a hybrid of a concatenation and a transformation-based data integration approach able to find similarities between samples across all platforms at once, following seven principal steps (See Figure). First, it maps features to the corresponding genes and uses a principal component analysis (PCA) per platform for an initial dimensionality reduction. Here we retained the 50 PCs with the highest eigenvalues for each platform. The contributions per platform are scaled on the total variance of each of their respective set of 50 PCs to ensure that the final integrated results are not dominated by a single platform. Next, the reduced features for each of the four platforms are concatenated resulting in a 200 dimensional space in which the samples are represented. Then, samples are mapped to a two dimensional multiplatform map using t-distributed stochastic neighborhood embedding (t-SNE). Here we used the Barnes-Hut implementation, an efficient implementation of t-SNE that handles thousands of samples. The power of the non-linear t-SNE mapping is that it retains the local similarity between samples instead of the relationships between dissimilar samples. The multiplatform map allows for subsequent analysis of the samples, such as cluster analysis, platform contribution analysis, or patient survival analysis.
Cite
Taskesen, E. et al., 2016. Pan-cancer subtyping in a 2D-map shows substructures that are driven by specific combinations of molecular characteristics. Scientific Reports, 6, p.24949. Available at: http://dx.doi.org/10.1038/srep24949
Software
The corresponding R package implementing MEREDITH is available at https://github.com/Kyoko-wtnb/meRedith
Complex Trait Genetics, VU Medical Center, Amsterdam
e.taskesen@vu.nl
Prof. Marcel J.T. Reinders
Pattern Recogntion and Bioinformatics, Delft University of Technology
m.j.t.reinders@tudelft.nl